En 1 clic






Surdités génétiques
Les surdités génétiques représentent plus des 50% des étiologies des surdités neurosensorielles. La découverte de l’implication croissante de mutations génétiques a modifié l’approche étiologique des ces surdités.
|
Rappel sur les modes de transmission génétique :
Autosomique récessif
- mode de transmission le plus fréquent (consanguinité)
- les 2 parents sont normo entendants mais tous deux porteurs de l’allèle atteint - 1 enfant sur 4 sera sourd - la présence de 2 allèles anormaux est indispensable pour qu’il y ait surdité
Autosomique dominant
- un des parents est sourd
- 1 enfant sur 2 sera sourd - l’enfant est atteint s’il est porteur d’un seul allèle « malade » - l’expressivité est variable selon la mutation dominante
Lié à l’X (récessif)
- sexe masculin (50% seront atteints)- transmission par les femmes
Lié à l'ADN mitochondrial
- transmission maternelle exclusive
- tous les enfants seront atteints (expressivité variable) |
Rappel sur la terminologie
Selon leur type de transmission, leur dénomination de surdités génétiques non syndromiques varie. Ainsi, les surdités appelées DFNB (de 1 à 20) évoquent une transmission autosomique récessive (AR). Une transmission autosomique dominante (AD) correspond à des surdités de type DFNA (1- 15). Enfin, les surdités liées à une transmission liée à l’X sont appelées DFN (1- 8).
- DFNA : pour locus autosomique dominant - DFNB : pour locus autosomique récessif - DFN : pour locus lié à l'X
DFNA3, DFNB1, ... (numérotation du locus) pour l'ordre chronologique de découverte de la localisation de la mutation génétique.
|
On divise les génosurdités en syndromiques et non syndromiques. La réalisation d’un questionnaire extrèmement précis, associé à un arbre généalogique et à un bon examen clinique ORL et pédiatrique avec un bilan para-clinique minimal permet de différencier les différentes étiologies des surdités.
Génosurdités syndromiques
Elles sont associées à un ou plusieurs autres symptômes cliniques et représentent environ 10 à 15 % des surdités de l’enfant. Elles représentent le tiers des génosurdités. Plus de 500 syndromes ont été décrits et plus de 100 gènes ont été identifiés à ce jour.
Les syndromes les plus fréquents doivent être recherchés par un bilan systématique. La surdité peut être le symptôme principal ou être au deuxième plan par rapport aux autres malformations.
Quelques syndromes sont à connaître:
Génosurdités non-syndromiques
Elles représentent 20 à 25 % des causes de surdités de l’enfant, soit les deux tiers des génosurdités, la surdité étant l’unique symptôme.
Probablement plus de 100 gènes sont en cause. Près de 70 gènes sont localisés et une trentaine de gènes différents est actuellement identifiée.
Les surdités autosomiques récessives
-
DFNB1 est la cause la plus fréquente des surdités de l'enfant ; elle est liée au gène de connexine.
Le gène est responsable à lui seul de 30 % des surdités de perception isolées qui surviennent dans l’enfance. La connexine est une protéine transmembranaire, composante de la jonction intercellulaire GAP permettant l’homéostasie de l’endolymphe.
La mutation la plus connue et la plus fréquente du gène de la connexine 26, localisée sur le chromosome 13, est celle de la 35delG, qui est en cause dans 50% des surdités autosomiques récessives dans les populations occidentales et du bassin méditerranéen. Le taux des porteurs sains de la mutation de la connexine 26 est évalué entre 2,5% et 4% de la population française. D’autres mutations sont affectées à différentes populations.
La surdité de perception est prélinguale le plus souvent, moyenne à profonde, bilatérale et symétrique, non peu ou pas évolutive. Il n’existe pas de retard à la marche. L'imagerie de l'oreille interne et les tests vestibulaires sont généralement normaux. Une mutation identique, mais de transmission dominante, a été retrouvée chez des enfants associant anomalies de la peau, kératite et surdité (KID syndrome). D’autres gènes de connexine peuvent être mutés et être à l’origine de surdité, telle la connexine 30.
- DFNB4 est une forme de surdité liée à une mutation du gène de la pendrine (PDS), également retrouvée dans le syndrome de Pendred. La surdité a les même caractéristiques; il existe une malformation de l’oreille interne (dilatation des aqueducs du vestibule), mais pas de pathologie thyroïdienne ; il n’existe donc pas de goitre et le bilan thyroïdien est normal.
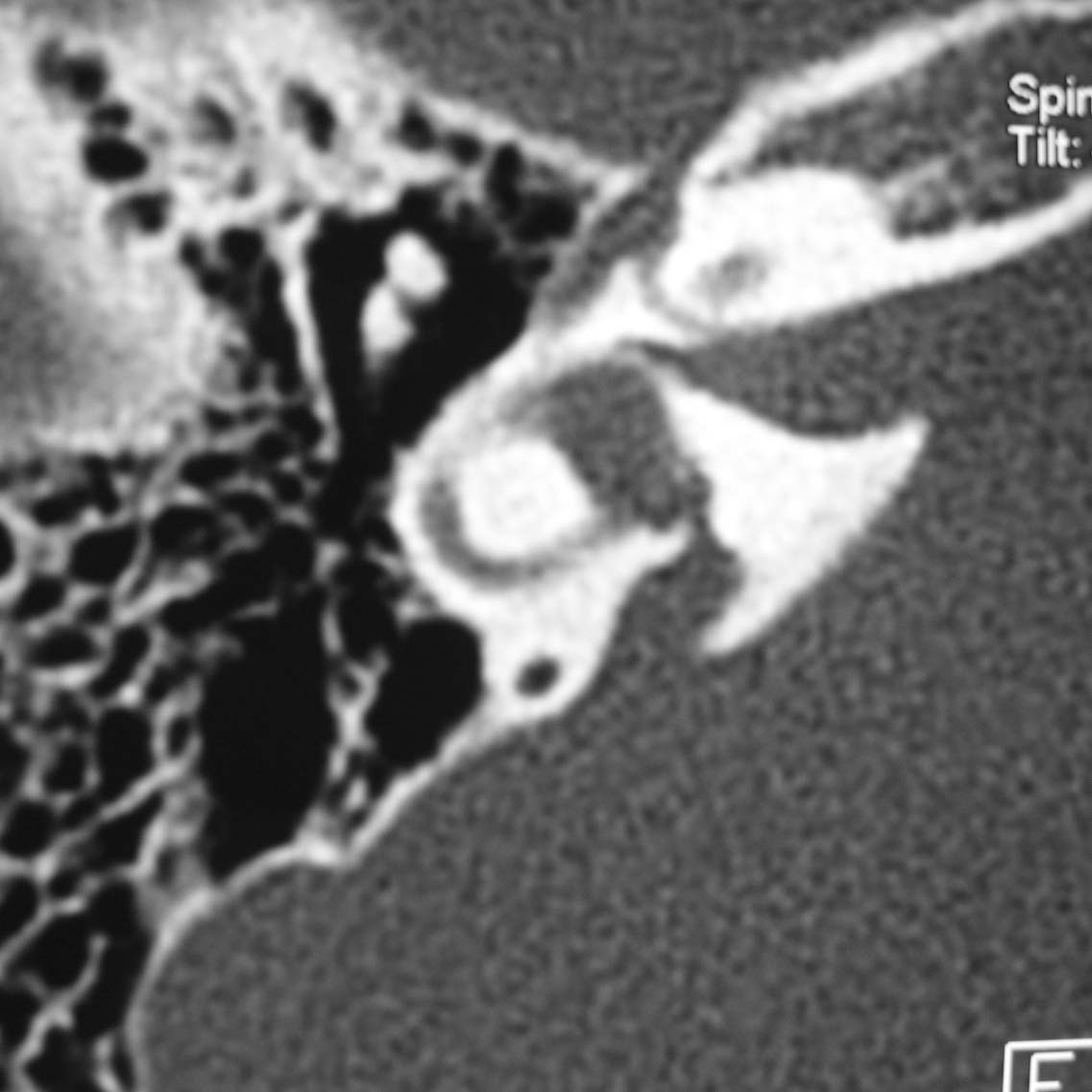
-
DFNB9 est une forme de surdité liée à une mutation du gène de l’otoferline (OTOF). Ce gène est normalement exprimé au niveau des cellules ciliées internes de l’organe de Corti. Cette protéine, l’otoferline, serait impliquée dans la fusion calcium-dépendante des vésicules synaptiques à la membrane plasmatiques des cellules sensorielles du vestibule et de la cochlée.
La surdité, habituellement sévère à profonde, a les caractéristiques d’une dyssynchronie auditive aux PEAP mais avec conservation des otoémissions acoustiques (au début tout au moins). Le scanner des rochers est normal. La consultation génétique et la pratique systématiques des OEA, en sus de PEAP pathologiques, doit faire rechercher cette mutation.
Les surdités autosomiques dominantes
- DFNA2 est une forme de surdité liée à une mutation du gène KCNQ4. Une mutation identique est présente chez les patients atteints du syndrome de Jervell et Lange-Nielsen. La surdité de perception est progressive touchant les fréquences aiguës.
- DFNA9 est une forme de surdité liée à une mutation du gène COCH. La surdité de perception est d’expression tardive (entre 15 et 65 ans) et de progression rapide.
La surdité est classiquement bilatérale, évolutive, associée à des acouphènes. Des signes vestibulaires à types d’instabilité ou de crises vertigineuses peuvent être associés. Le scanner des rochers est normal. Le bilan vestibulaire objective une hypo ou une aréflexie. Le principal diagnostic différentiel est la maladie de Ménière.
- DFNA13 est une forme de surdité liée à une mutation du gène COL11A2. La surdité de perception est stable, touchant les fréquences moyennes.
- DFNA6/DFNA14/DFNA38 regroupe la surdité des basses fréquences. Elle se caractérise par une atteinte de l’audition prédominant sur les basses fréquences ( < 2000 Hz). La surdité apparait dans l’enfance et est lentement progressive. Des mutations du gène WFS1 ont été identifiées.
Les surdités liées au chromosome X
-
DFN3 est une forme de surdité liée à une mutation du gène POU3F4. On la nomme le « Geyser labyrinthe » ou pseudo-otospongiose. La surdité est isolée, souvent à composante mixte, de degré variable (moyenne à sévère). Néanmoins, elle peut s’associer à des troubles vestibulaires.
Elle simule une malformation d’oreille moyenne de type blocage ossiculaire. La TDM des rochers est indispensable pour retrouver une dilatation de la cochlée, du vestibule et du conduit auditif interne et empêcher à l’occasion d’une exploration d’oreille un geste de type stapédectomie responsable d’un geyser labyrinthe avec risque de cophose et /ou de méningite. Tout geste platinaire s’accompagne d’une Geyser avec risque important de cophose imposant la réalisation d’un scanner avant tout geste chirurgical de l’enfant.
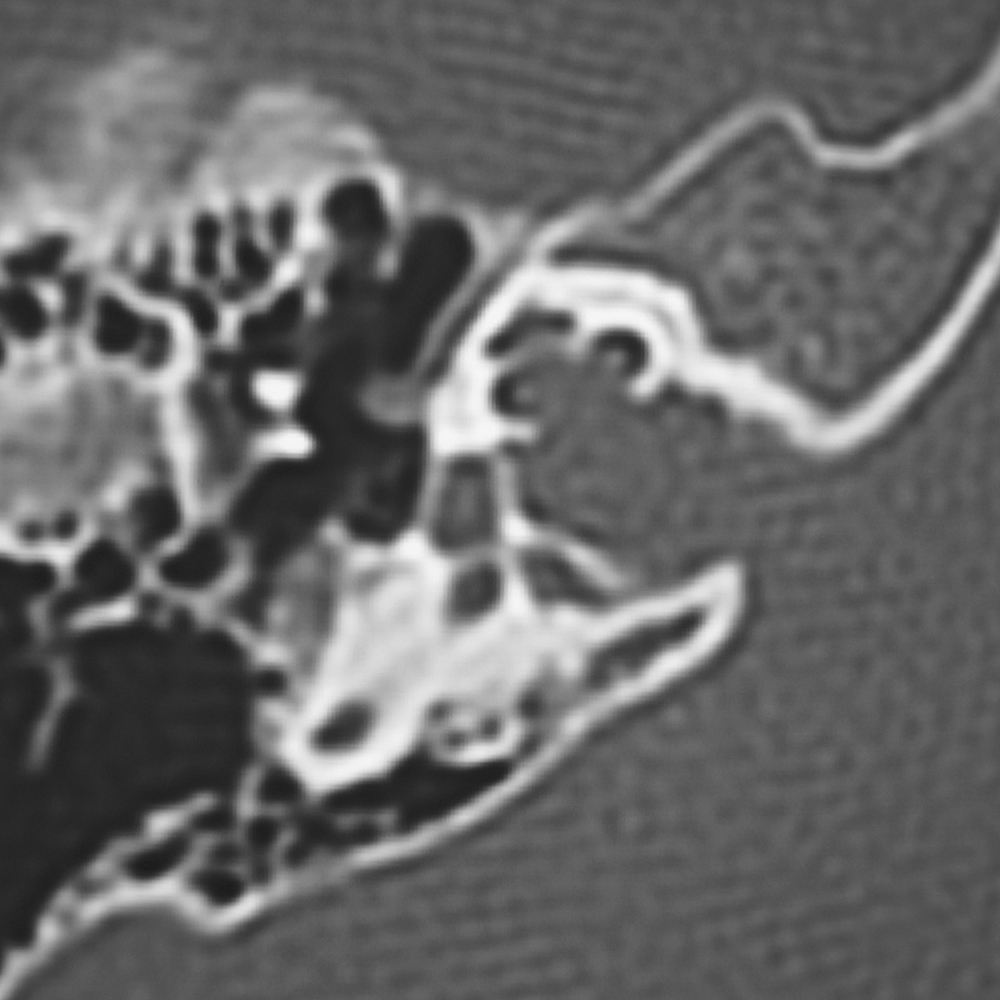
Les surdités mitochondriales
Les mutations responsables de la surdité sont localisées dans l’ADN mitochondrial. Ces mutations sont essentiellement transmises par la mère à l’ensemble de leurs enfants. L’expressivité de ces mutations est variable, pouvant aussi donner diverses maladies touchant des organes qui sont de grand consommateur d’énergie (cœur, cerveau, rétine, muscle, pancréas). Les signes cliniques, autres que la surdité, sont extrêmement variés: neuropathies, cardiopathies, diabète, dégénérescence rétinienne, …
Actuellement, 8 mutations mitochondriales responsables de surdité neurosensorielles isolées ont été identifiées. La surdité due à la mutation A1555G est reconnue à l’origine d’une susceptibilité de la cochlée de certains patients aux antibiotiques aminosides. La surdité de perception est de sévérité variable et d’évolution progressive.
De nombreuses mutations mitochondriales sont également retrouvées dans certaines génosurdités syndromiques (MELAS, MERRF, MNGIE).
QUELQUES SYNDROMES
| Waardenburg | Usher | Pendred | BOR |
| Jervell et Lange-Nielsen | Alport | Stickler |
Le syndrome de Waardenburg
Il représente 2-5% des surdités congénitales. La fréquence de ce syndrome est certainement sous-estimée. En effet, les signes cliniques peuvent passer inaperçus (écartement de canthi de façon isolée sans surdité ni autre anomalie dans le type II). Il existe une variabilité importante de l’expressivité. Le rendement du diagnostic moléculaire est encore faible. La transmission est autosomique dominante. Le gène responsable du type I est le gène PAX3. Le métabolisme de la mélanine serait ainsi altéré. La résultante de cette anomalie est donc une absence de mélanocyte Il existe 4 types cliniques de ce syndrome, causés par des mutations retrouvées dans 7 gènes différents.
Le diagnostic de syndrome de Waardenburg est posé si l’enfant présente 2 critères MAJEURS, ou 1 critère MAJEUR associés à 2 critères mineurs.
|
Les critères MAJEURS sont :
- Apparenté du 1 er degré atteint de SW - Présence d’une dystopie des canthi et des cheveux blancs avant l’âge de 30 ans (pour SW de type II) - Présence d’une mèche blanche - Surdité de perception - Anomalie de l’iris Les critères MINEURS sont : - Existence de zones cutanées dépigmentées - Une racine du nez large et haute - La partie médiane des sourcils épaisse ou synophrys - Une hypoplasie des ailes du nez - Des cheveux blancs avant l’âge de 30 ans |
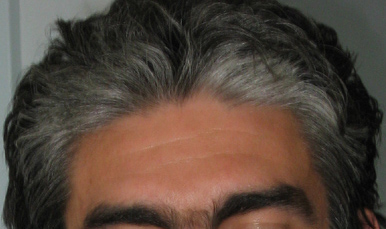
La surdité est congénitale, neurosensorielle, de degré variable. Elle est profonde dans 35 % des cas. Elle est le plus souvent bilatérale et peut être progressive. 4 types peuvent être distingués :
- Waardenburg de type I (SW1) : le plus fréquent - dystopie canthale.
- Waardenburg de type II (SWII) : Sa fréquence est probablement sous estimée, en raison de la pauvreté de ses signes cliniques qui peuvent passer facilement inaperçus. Les anomalies radiologiques sont fréquentes.
- Waardenburg de type III (syndrome de Klein Waardenburg, SWIII) : Le phénotype est identique au SW1 mais les patients présentent en plus, une hypoplasie des membres supérieurs associée.
- Waardenburg de type IV (syndrome de Shah Waardenburg, SWIV) : maladie d’Hirschprung associée aux autres signes cliniques déjà cités
Le fond d’œil, en objectivant une dépigmentation, peut aider au diagnostic. Les tests vestibulaires caloriques peuvent être perturbés d’une façon uni-ou bilatérale. Le scanner des rochers peut mettre en évidence des anomalies radiologiques tels qu’un élargissement du sac endo-lymphatique (sans réelle dilatation de ce sac), une dysplasie vestibulaire (une aplasie des canaux semi circulaires est même possible). Un peu moins de 50 % des enfants atteints présenteraient des anomalies radiologiques
Le syndrome d’Usher
Il est, à lui seul, responsable de 2 à 10 % des surdités congénitales et 18 % des rétinopathies pigmentaires. Ce syndrome est la cause la plus fréquente de surdité associée à une cécité.
La transmission est autosomique récessive. La mutation se situe sur le chromosome 13, au niveau du locus du gène codant pour la myosine. six gènes sont actuellement identifiés.
Classiquement, l’enfant, atteint d’un syndrome d’Usher présentera une surdité congénitale et une rétinite pigmentaire (RP) dont le pronostic visuel est la cécité. La précocité et la sévérité des atteintes auditives et rétiniennes, ainsi que l’existence d’une anomalie vestibulaire associée permettent de différencier les différentes formes cliniques de ce syndrome. Le diagnostic positif est souvent tardif en raison de l’hétérogénéité clinique. La surdité est classiquement de sévérité moyenne à profonde, tardive et progressive.
Trois formes cliniques se différentient.
- Le Type I est le plus fréquent et le plus sévère. Ces enfants présentent une surdité profonde pré-linguale bilatérale congénitale, associée à une aréflexie vestibulaire congénitale bilatérale responsable d’un retard des acquisitions motrices pendant les premières années de vie. Classiquement, la RP apparait de façon précoce en période pré-pubertaire (vers l’âge de 10 ans). Elle est responsable d’une cécité nocturne précoce dans l’enfance, de scotomes à l’adolescence et d’une cécité totale chez le jeune adulte.
- Le Type II : Ces enfants présentent une surdité moyenne à sévère, stable et d’apparition tardive. La perte auditive est prédominante dans les hautes fréquences. Elle reste modérée à légère dans les basses fréquences. Ces enfants ne présentent pas de troubles vestibulaires. La rétinite pigmentaire apparait tardivement et est cliniquement symptomatique vers l’âge de 20 à 30 ans. Son évolution est moins sévère que dans le type I. Elle est responsable d’une cécité nocturne à l’adolescence, de scotomes vers 20 ans et d’une cécité totale vers 30 ou 40 ans.
- Le Type III : La surdité débute à la puberté. Elle est dite tardive et évolutive, et est plus ou moins associée à une atteinte vestibulaire. La RP apparait plus tardivement que dans le type I et est moins sévère : la cécité nocturne précède systématiquement la cécité totale. Cette forme clinique semble particulièrement fréquente dans les pays nordiques.
| La rétinite pigmentaire se définit par une baisse de la nyctalopie (c'est-à-dire une baisse de l’acuité visuelle nocturne) dont les signes précoces sont une baisse de l’acuité visuelle progressive, un rétrécissement du champ visuel, et une anomalie de la vision des couleurs. Elle est asymptomatique et inapparente pendant la première décennie (excepté pour le syndrome de Usher de type I). |
Concernant le bilan complémentaire, un TDM des rochers sera sans anomalie. Le fond d’œil est le plus souvent normal. Dans le Usher de type I, les premiers signes apparaissent vers 13-14 ans. La normalité du fond d’œil n’élimine en aucun cas un syndrome d’Usher. L’électrorétinogramme (ERG) est fondamental dans le diagnostic. Il est pathologique précocement. Un ERG normal avant l’âge de 3 ans, ne peut éliminer formellement le diagnostic de syndrome d’Usher. Il doit être réitérer 6 à 12 mois plus tard. Le diagnostic moléculaire ne se fait pas en routine. L’hétérogénéité clinique complique le diagnostic. Plusieurs gènes sont impliqués non seulement pour chacun des trois types mais aussi pour un même type de syndrome d’Usher.
Le traitement préventif (à type de prévention secondaire) prend toute son importance dans ce syndrome. Il est important de dépister précocement ces enfants présentant un syndrome d’Usher car une rééducation orthophonique avec une pose d’implant cochléaire (dans le cadre d’une surdité sévère à profonde bilatérale) permettra de stimuler l’enfant, de façon optimale, alors qu’il sera confronté à un double handicap. L’Implant cochléaire doit être réalisé rapidement, que la rétinite pigmentaire survienne précocement ou non. C’est une des indications en urgence d’implantation cochléaire bilatérale.
Le syndrome de Pendred
Sa prévalence est de 4 à 10 % parmi les sujets atteints de surdité congénitale sévère à profonde. C’est probablement une des plus fréquentes causes de surdité syndromique pré-linguale. La transmission est autosomique récessive. La mutation responsable de ce syndrome se situe au niveau du gène PDS qui code pour une protéine, la pendrine. La pendrine est exprimée au niveau de la thyroïde, de l’oreille interne et du rein et joue un rôle de transporteur d’iode, de chlore. Sa présence au sein des régions cochléaires où s’effectue la résorption du liquide endolymphatique nous fait supposer qu’elle intervient dans l’homéostasie des liquides de l’oreille interne. Une altération de ce transporteur entraîne un trouble au niveau thyroïdien, du transport et de l’organification de l’iode (trouble de l’hormonogenèse à l’origine du goître), au niveau de l’oreille interne une anomalie de résorption du liquide endolymphatique (à l’origine d’une surdité). Aucune anomalie rénale n’a été décrite à ce jour.
Ce syndrome associe :
- Une surdité qui est de degré variable (de légère à profonde), bilatérale prédominant sur les fréquences aigues. Elle est congénitale ou post-linguale précoce, évolutive (progressive dans 15 % des cas ou fluctuante avec possibles surdités brusques à répétition). Elle peut être isolée ou associée à un goître thyroïdien.
- Un trouble des fonctions vestibulaires, observable chez la plupart des malades
- Un goitre qui peut être congénital ou apparaîssant vers la fin de la première décennie. Il est présent avant la puberté dans 40 % des cas (mais est rarement présent avant 10 ans) ou apparaît à l’âge adulte dans 60 % des cas. L’enfant est euthyroïdien dans 50 % des cas ou hypothyroïdien dans 50 % des cas.
Concernant le bilan complémentaire, une TDM des rochers sera prescrite et sera pathologique dans 85 % des cas (dilatation des aqueducs du vestibule, malformation de Mondini, hypoplasie cochléaire. Le bilan thyroïdien retrouve un dosage des hormones thyroïdiennes normal (mutation du gène DFNB4) ou augmenté dans 10 % des cas. Le test au Perchlorate, précisant l’anomalie d’organification de l’iode, étudie la sécrétion d’iode radioactif dans les 2 heures qui suivent l’administration intraveineuse de Perchlorate. Une sécrétion supérieure à 10 % est « évocatrice » du syndrome, en raison du manque de spécificité et sensibilité du test. Avant l’âge de 10 ans, il peut être le seul test à être perturbé et on a ainsi une diminution de la fixation thyroïdienne de l’iode radioactif, après administration de Perchlorate ou de Thiocyanate. L’échographie thyroïdienne peut être normale ou peut objectiver un goitre. L’étude génétique peut mettre en évidence différentes mutations.
Le traitement préventif consiste en la vaccination anti-pneumococcique. Il est nécessaire d’éviter les sports violents et les traumatismes pressionnels et d’essayer de limiter les traitements par corticoïdes pour les épisodes de surdités brusques. Chez les patients euthyroïdiens, une supplémentation en iode permet de réduire le goître. Quand une hypothyroïdie existe, le traitement curatif est la L-thyroxine ; celui-ci peut prévenir l’apparition du goitre. Aucun traitement médicamenteux ne permet d’enrayer l’évolution ou de restaurer l’audition.
| Syndrome de Pendred | Surdité DFNB4 |
|
|
|
|
|
|
|
|
Le syndrome Branchio-Oto-Rénal (BOR)
Il représente 2 % des surdités congénitales. Son mode de transmission est autosomique dominant. Un gène est actuellement identifié (mutation sur le gène EYA1 situé sur le chromosome 8 pour le type 1 et mutation sur le gène SIX5 pour le type 2)
L’expressivité est variable: le diagnostic positif repose sur un faisceau d’argument clinique. Les signes du syndrome sont souvent retrouvés dissociés dans la famille. Les signes du BOR sont parfois discrets. La recherche des antécédents familiaux, dans cette pathologie autosomique dominante est fondamentale.
Il faut rechercher :
- des anomalies des arcs branchiaux : fistules préhélicéennes, de la 2ème fente, résidus branchiaux cartilagineux plutôt bilatéraux
- des anomalies de l’oreille externe : pavillon malformé, conduit auditif externe étroit, enchondromes , oreilles petites et antéversée…
- des malformations rénales : uni ou bilatérales, de sévérité variable allant d’un rein de petite taille ou malformé jusqu’aux hypoplasies majeures et agénésies
La surdité peut être de perception (30 % des cas), de type mixte (50 %) ou transmissionnelle (30%). Quand elle est neurosensorielle, la surdité peut être congénitale ou d’apparition progressive.
 |
 |
 |
|
Anomalies otologiques
|
Anomalies branchiales – fistules préhélicéennes
|
Anomalies branchiales – fistule du 2ème arc
|
Parmi les examens complémentaires :
- l’échographie rénale recherchera des malformations rénales (hypoplasie rénale, rein enfer à cheval, agénésie rénale, duplication pyélique, défaut de rotation …)
- le scanner des rochers présentera classiquement comme anomalies un tour apical cochléaire hypoplasique, une déviation médiale du VII, un élargissement du CAI ou de la trompe d’eustache.
- les tests génétiques sont couteux et peu accessibles en routine. Seulement 40 % des BOR ont une mutation du gène EYA1.
Le syndrome de Jervell et Lange-Nielsen
Il reste rare mais le prognostic vital d’un enfant peut être engagé s’il est méconnu. Il représente 0,1 % à 1 % des surdités congénitales. 6 enfants sur 1000 enfants sourds présentent ce syndrome. Sa transmission est autosomique récessive. Deux gènes sont actuellement identifiés : gènes majoritairement exprimés dans la strie vasculaire. Leur mutation provoque donc des anomalies au niveau des cellules sécrétant l’endolymphe. Les canaux potassiques des cellules myocardiques sont aussi altérés.
La surdité est profonde ou sévère, bilatérale, prédominante sur les fréquences aigues, congénitale et isolée. Il n’existe pas d’anomalie vestibulaire. L’électrocardiogramme peut mettre en évidence un trouble de la conduction cardiaque à type d’allongement de l’espace QT ou des troubles de la repolarisation. Ces anomalies sont à l’origine de syncopes survenant lors d’un stress (typiquement, syncope lors des pleurs ou des jeux) mais elles peuvent aussi conduire à la mort subite. Du fait de la transmission autosomique récessive, la présence d’antécédents familiaux n’est pas classique mais devant la possible gravité de ce syndrome, il est légitime de rechercher à l’interrogatoire la présence de malaises ou de morts subites dans la famille.
Les examens para-cliniques à réaliser sont l’électrocardiogramme (allongement de l’espace QT…), une échographie trans-thoracique (cœur de morphologie normale, avec une bonne fonction ventriculaire), un scanner des rochers (normal) et un bilan sanguin ( possible anémie hypochrome).
Chez les patients présentant un syndrome de JLN, le pronostic vital est engagé. Malheureusement, le diagnostic de ce syndrome est souvent retardé, exposant les enfants au risque de mort subite par arythmie ou asystolie. Environ, un enfant sur deux, atteint de ce syndrome meure avant l’âge de 20 ans. Dans la littérature, on retrouve que 15 % des enfants deviennent symptomatiques avant l’âge de 23 mois. A partir de 18 ans, au moins 90 % des patients présentant un syndrome de JLN ont présenté un évènement cardiaque. Il est nécessaire de prescrire de façon systématique un ECG à tous les enfants présentant une surdité sévère à profonde non étiquetée, surtout si ces derniers doivent bénéficier d’une anesthésie générale.
Quand un syndrome de JLN est avéré, une prévention des facteurs de stress doit être faite et des précautions doivent être prises pendant un acte chirurgical. Les enfants présentant un syndrome de JLN doivent être traités par Béta-bloquants. Ces derniers diminuent le risque de mortalité de > 50 % à 5 %.
Le syndrome d’Alport
Il représente 1 % des surdités congénitales. Son mode de transmission est dominant lié au chromosome X dans 85 % des cas. Trois gènes sont actuellement identifiés.
La surdité est due à une atteinte du collagène de type IV au niveau de sa chaine alpha, située principalement au niveau des membranes basales des glomérules rénaux, de l’œil, de l’oreille interne, des plexus choroïdiens et du poumon. Ces membranes dégénèrent principalement au niveau de la strie vasculaire cochléaire et du glomérule rénal.
La surdité est précoce et prédomine sur les fréquences aigües dans la première décennie. Elle est classiquement bilatérale, progressive et asymétrique. Des hématuries et protéinuries sont souvent présentes mais latentes car microscopiques ; c’est la glomérulonéphrite microscopique. Des anomalies oculaires sont possibles de type cataracte, kératocône et rétinite pigmentaire.
Classiquement, la surdité et l’atteinte oculaire apparaissent après l’atteinte rénale.
Le bilan para-clinique se compose d’un fond d’œil (lenticone) et d’une bandelette urinaire (hématurie microscopique puis macroscopique puis insuffisance rénale). Sur le plan vestibulaire, on note une hypo réflexivité.
L’évolution est bénigne chez la femme. Elle peut être grave chez l’homme allant jusqu’à l’insuffisance rénale terminale. Ce syndrome représente 2 % des insuffisances rénales terminales.
Les atteintes rénales, cochléaires et ophtalmologiques évoluent dans ce syndrome de façon parallèle.
Le syndrome de Stickler
Il représente 0,25 % des surdités congénitales. Son mode de transmission est autosomique dominant ; trois gènes sont actuellement identifiés. L’expressivité est variable : les signes du syndrome sont souvent retrouvés dissociés dans la famille. Il faut rechercher :
- une fente palatine
- une myopie
- des anomalies squelettiques, cartilagineuses et ligamentaires (hyperlaxité). Les patients atteints sont souvent grands et minces, de type marfanoïde.
On peut retrouver également des décollements de rétine (favorisées par la forte myopie), une cataracte, une micrognathie, une arthrose précoce.
Concernant la surdité, elle est neurosensorielle stricte dans 80 % des cas mais elle peut être mixte (probablement à causes d’otites séro-muqueuses favorisées par la fente palatine). Quand elle est neurosensorielle, elle est sévère à profonde, évolutive.
Parmi les examens complémentaires, l’examen ophtalmologique prend toute sa place.