En 1 clic






État de santé des enfants atteints de bêta-thalassémie en France, un pays à revenu élevé
Le centre de référence de Marseille pour les Syndromes Drépanocytaires Majeurs, Thalassémies et autres Pathologies rares du Globule rouge et de l’Erythropoïèse (Dr Isabelle Thuret) vient de publier un rapport sur l’état de santé des enfants atteints de bêta-thalassémies en France, un pays à revenu élevé. Il est issu des travaux de thèse de Dr Caroline Donzé, thèse soutenue à Marseille en septembre dernier, qui visait à décrire l'épidémiologie et les complications des formes sévères (majeures et intermédiaires) de la bêta-thalassémie en pédiatrie.
La bêta-thalassémie est une maladie génétique rare en France caractérisée par un défaut de synthèse de la chaîne de bêta-globine. Dans sa forme la plus sévère (majeure), elle entraîne une anémie devant être corrigée par transfusions sanguines dès la petite enfance. Grâce à l’amélioration des traitements chélateurs du fer (pour prévenir la surcharge en fer secondaire aux transfusions), elle est devenue une maladie chronique avec une médiane de survie d’environ 50 ans (Lien Autres ressources sur le site de la filière MCGRE).
Le travail mené par le Dr C Donzé sous la direction du Dr Valentine Brousse s’est basé sur le recueil des données du Registre National des Thalassémies (NaThalY)* animé par le Pr Catherine Badens. Il a inclus 237 patients nés entre 2005 et 2020 (âge médian de 7.1 ans lors de la dernière visite), atteints d’une β-thalassémie intermédiaire (TI) ou majeure (TM, ~2/3 des patients). Ces patients avaient donc eu accès à la chélation orale en fer par le déférasirox, qui a été commercialisé en 2007. Mettant en lumière les caractéristiques de cette cohorte française et les pratiques de prise en charge, les résultats confirment le bon pronostic déjà observé dans d’autres pays riches :
- Bêta-Thalassemia in childhood: Current state of health in a high-income country. Par Donze C, Benoit A, Thuret I, Faust C; le « NaThalY Network », Gauthier A, Berbis J, Badens C, Brousse V. Br J Haematol. 2023. https://doi.org/10.1111/bjh.18631. Lien PubMed
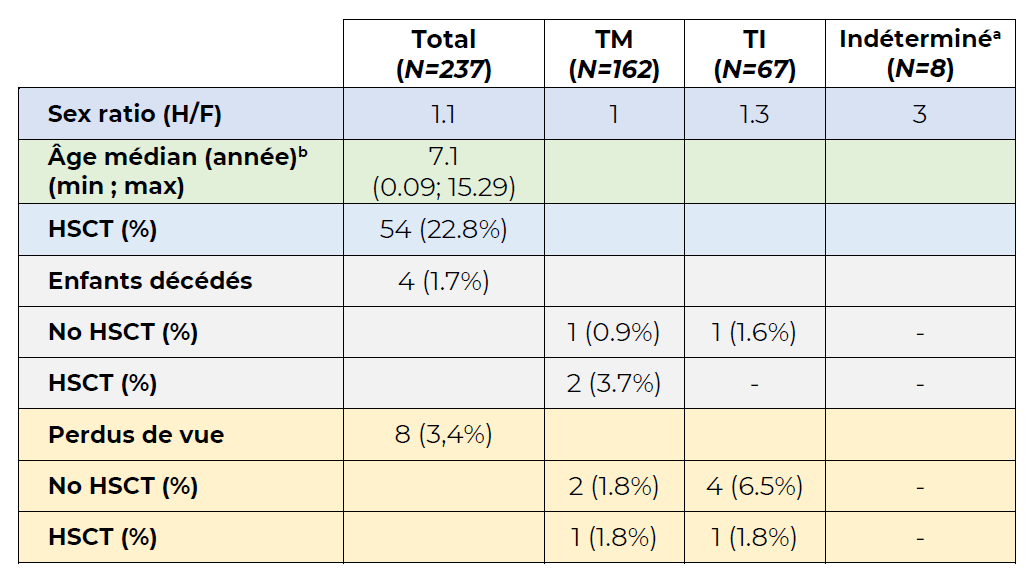
Table- Caractéristiques cliniques de la population étudiée - Table simplifiée extraite de la Table 1 de l’article par Donze C et al. Br J Haematol. 2023 (Lien PubMed)
Abréviations :
HSCT, transplantation de cellules souches hématopoïétiques
TI, bêta-thalassémie intermédiaire
TM, bêta-thalassémie majeure
aLes patients étaient trop jeunes pour la classification en TI ou TM
bÂge auquel la moitié de la population a eu sa dernière visite dans une équipe hospitalière française prenant en charge la bêta-thalassémie