En 1 clic






Cohorte française de patients atteints de la forme infantile classique de la maladie de Pompe : suivi à long terme de 64 enfants
Le suivi jusqu’à 17 ans de la cohorte française de patients atteints de la forme infantile classique de la maladie de Pompe (MP) vient d’être publié dans la revue médicale European Journal of Neurology. Ce travail collaboratif a été porté par le Pr Brigitte Chabrol avec la chef de projet Céline Cudejko (Centre de Référence Coordonnateur des Maladies Héréditaires du Métabolisme CoMMet) à Marseille et a été mené en étroite collaboration avec les CRMR prenant en charge les patients, en particulier le CRMR maladies héréditaires du métabolisme de Tours ToTeM.
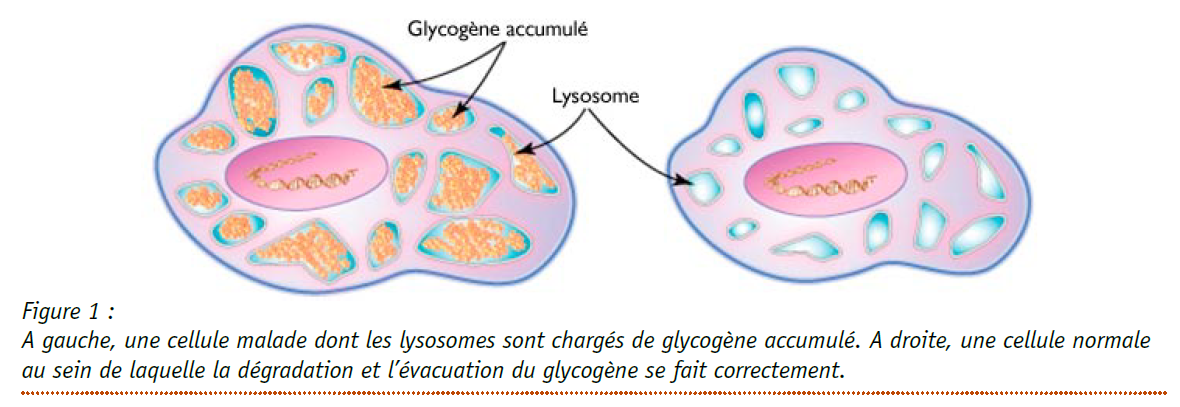
La Maladie de Pompe est une maladie de surcharge lysosomale à transmission autosomique récessive due à un déficit en alpha-glucosidase acide (GAA) ou maltase acide. Egalement appelée glycogénose de type 2, elle provoque une accumulation intra-lysosomale de glycogène dans la plupart des tissus, mais les symptômes sont principalement dus à l’altération des muscles squelettique et cardiaque. La forme infantile classique est la forme la plus sévère. Elle se manifeste avant l’âge de 6 mois (voire dès la période anténatale) par une cardiomyopathie hypertrophique, une hypotonie et des troubles respiratoires sévères avec un décès précoce en l’absence de traitement, habituellement avant l’âge de 12 mois.
Depuis 2004 il existe un traitement spécifique : l’alglucosidase alfa (Myozyme®). C’est un traitement enzymatique substitutif (enzyme replacement therapy ou ERT), qui a considérablement augmenté le taux de survie des patients.
Dans cette étude, les auteurs ont analysé rétrospectivement l’évolution de 64 patients atteints de la forme infantile classique de la MP, diagnostiqués en France entre 2004 et 2020. Pour se faire, un recueil de données anonymisé a été réalisé auprès des centres experts des filières G2M et Filnemus. Les données ont été collectées, centralisées puis analysées par l’équipe du CRMR CoMMet.
Merci aux auteurs pour ce rapport sur l’une des plus grandes cohortes et sur le plus long suivi des patients atteints de cette maladie. Il met en exergue que l'ERT a changé l’histoire naturelle de la maladie, cependant elle reste associée à un risque augmenté de mortalité et souvent à un mauvais pronostic. Cette modalité de traitement ne permet pas d’éviter l'évolution, à moyen terme, de la maladie vers une pathologie neuromusculaire et du système nerveux central, ce qui renforce la nécessité de développer de nouvelles thérapies.
-
Long-term follow-up of 64 children with classical infantile-onset Pompe disease since 2004: A French real-life observational studyPar Marine Tardieu, Céline Cudejko, Aline Cano, Célia Hoebeke, Delphine Bernoux, Violette Goetz, Samia Pichard, Anaïs Brassier, Manuel Schiff, François Feillet, Paul Rollier, Karine Mention, Dries Dobbelaere, Alain Fouilhoux, Caroline Espil-Taris, Didier Eyer, Frédéric Huet, Ulrike Walther-Louvier, Magalie Barth, Laurent Chevret, Alice Kuster, Jérémie Lefranc, Julien Neveu, Gaele Pitelet, Juliette Ropars, François Rivier, Agathe Roubertie, Guy Touati, Catherine Vanhulle, Emilie Tardieu, Catherine Caillaud, Roseline Froissart, Murielle Champeaux, François Labarthe, Brigitte Chabrol.Eur J Neurol. 2023 May 26. doi: 10.1111/ene.15894. Lien article
- En savoir plus sur la Maladie de Pompe
* Lien Orphanet _ La Maladie de Pompe
Figure tirée de la Fiche Orphanet La Maladie de Pompe-2010 :
* Maladie de Pompe_ AFM Téléthon