En 1 clic






Pistes thérapeutiques pour la maladie de Charcot Marie-Tooth (CMT)
La maladie de Charcot-Marie-Tooth (CMT) regroupe un ensemble hétérogène de neuropathies motrices et sensorielles, qui ont une cause génétique. Chaque type de CMT est rare, mais l’ensemble représente les neuropathies héréditaires les plus fréquentes au monde.
Les symptômes débutent le plus souvent à l’adolescence, les anomalies génétiques entraînant une atteinte des nerfs périphériques, en particulier ceux des jambes et des bras. Le handicap associé est en général lentement progressif (cf. lien Maladie de Charcot-Marie-Tooth AFMTÉLÉTHON). Mais les manifestations cliniques et la gravité dépendent, notamment, du gène muté impliqué.
Dans une synthèse récemment parue dans la « Revue neurologique », internationale, le Pr Shahram Attarian- coordonnateur du CRMR Maladies Neuromusculaires Rares - PACA Réunion Rhône-Alpes et Sadia Beloribi-Djefalia- chef de projets, offrent un état de l’art des thérapies pour ces pathologies (1).
En fait, il n'y a pas encore de traitement disponible permettant de les guérir. La prise en charge des patients repose sur la rééducation, l'ergothérapie*, la réduction de la fatigue et de la douleur, parfois de la dépression. Le suivi régulier des patients permet d’adapter la prise en charge des patients à leurs besoins et à la sévérité de la maladie.
Mais au cours de ces 30 dernières années, la recherche a mené à des progrès importants sur l’identification des gènes impliqués dans les différents types de CMT, et des voies de signalisation en cause : au niveau des cellules musculaires, des neurones et des cellules de Schwann (cf. liens Cellules de Schwann AFMTÉLÉTHON).
En particulier, l'équipe du CRMR coordonnateur Maladies Neuromusculaires Rares - PACA Réunion Rhône-Alpes (Pr Shahram Attarian) avec ses internes a récemment apporté à la communauté internationale de nouvelles données sur les relations entre le génotype/phénotype moléculaire et le handicap (2).
Grâce à ces caractérisations moléculaires, il est envisageable de concevoir de nouvelles stratégies thérapeutiques spécifiques aux pathologies : thérapies géniques, nouveaux inhibiteurs et modulateurs. C’est en ce moment charnière que la revue vous décrit les essais cliniques en cours, et les perspectives : pour les formes principales et les formes rares de CMT (Figures 1&2) (1).
Les recommandations de prise en charge, coordonnée, multidisciplinaire, sont aussi explicitées par le centre de référence ; elles adressent notamment la chirurgie orthopédique, et l’éducation thérapeutique du patient et de sa famille (pour l’adaptation du patient à sa maladie chronique).
Ce guide représente une référence pour les neurologues et autres professionnels prenant part au soin de ces patients, les chercheurs et tout public intéressé.
* « Occupational therapy », qui vise à maintenir, restaurer et faciliter les activités de la vie quotidienne de manière sécurisée, autonome et efficace.
-
(1) Treatment of Charcot-Marie-Tooth neuropathies par Beloribi-Djefaflia S & Attarian S. Revue Neurologique (Paris). 2023. https://doi.org/10.1016/j.neurol.2022.11.006
-
(2) Série d’articles réalisés par les internes du CRMR Maladies neuromusculaires rares de Marseille (Pr S Attarian), avec les équipes hospitalières françaises du réseau du centre. Les données sont issues de patients atteints de CMT suivis dans ces équipes.
Genotype–phenotype correlation in French patients with myelin protein zero gene-related inherited neuropathy par Subréville M, Bonello-Palot N, Yahiaoui D, Beloribi-Djefaflia S, Fernandes S, Stojkovic T, Cassereau J, Péréon Y, Echaniz-Laguna A, Violleau MH, Soulages A, Léonard-Louis S, Masingue M, Magot A, Delmont E, Sacconi S, Adams D, Labeyrie C, Genestet S, Noury JB, Chanson JB, Lévy N, Juntas-Morales R, Tard C, Sole G, Attarian A. Eur J Neurol. 2021;28:2913–2921. https://doi.org/10.1111/ene.14948
-> A permis de formuler des recommandations sur les critères d’inclusion dans les essais cliniques testant des inhibiteurs de réponse aux protéines non repliées (UPR) (tels que Sephin1), chez les patients atteints de CMT, porteurs d’une mutation dans le gène MPZ (qui code la protéine de la myéline Po) :
- Il faudrait tenir compte des patients chez qui UPR est activée
- Et inclure les patients présentant une gravité légère à modérée de la maladie et un début de maladie entre 18 et 50 ans. Ceux-ci sont en effet sujets à une progression significative de la maladie au fil du temps.
Fat fraction distribution in lower limb muscles of patients with CMT1A: A quantitative MRI study par Bas J, Ogier AC, Le Troter A, Delmont E, Leporq B, Pini L, Guye M, Parlanti A, Lefebvre MN, Bendahan D, Attarian S. Neurology. 2020.
-> Etude réalisée sur des personnes atteintes de CMT1 suivies dans les centres français et de volontaires non atteints (contrôles).
-> A mis en évidence une infiltration graisseuse musculaire typique chez les patients CMT1A, corrélée à des déficits cliniques (notamment de la force musculaire).
Motor unit number index correlates with disability in Charcot-Marie-Tooth disease.
par Bas J, Delmont E, Fatehi F, Salort-Campana E, Verschueren A, Pouget J, Lefebvre MN, Grapperon AM, Attarian S. Clin Neurophysiol. 2018.
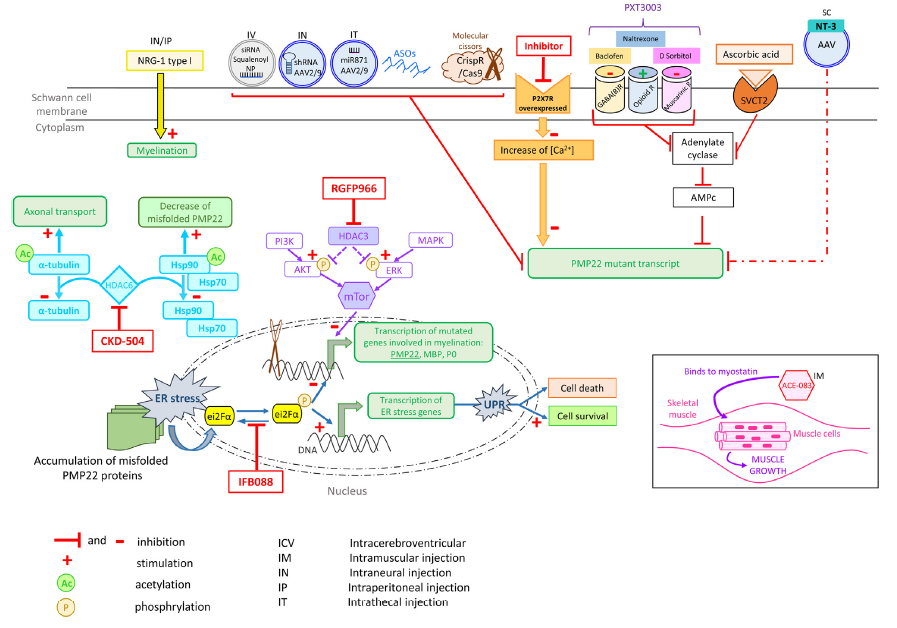
Figure 1_tirée de la revue de Beloribi-Djefaflia S & Attarian S. Treatment of Charcot-Marie-Tooth neuropathies. 2023 :
Targeting CMT1A. Schematic representation of currently tested drugs to slow down CMT1A disease progression, whether at preclinical or clinical stages
Représentation schématique des médicaments actuellement testés pour ralentir la progression de la CMT1A, que ce soit au stade préclinique ou clinique.
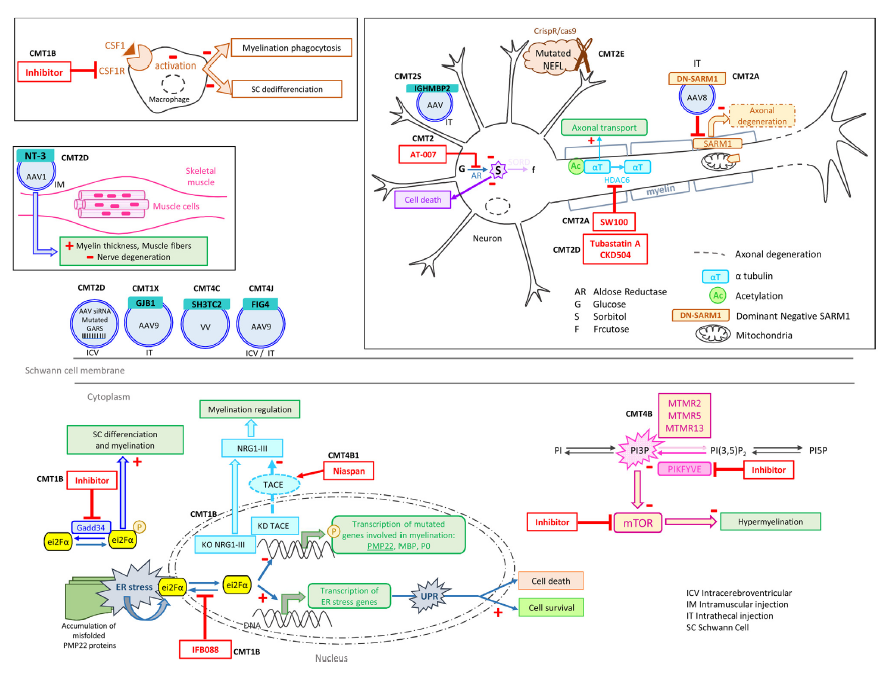
Figure 2_tirée de la revue de Beloribi-Djefaflia S & Attarian S. Treatment of Charcot-Marie-Tooth neuropathies. 2023 :
Targeting other forms of CMT. Schematic representation of targeted pathways in other types of CMT.
Ciblage d'autres formes de CMT. Représentation schématique des voies ciblées dans d'autres types de CMT.